
Болезнь бабочки или беллезный эпидермолиз. Что это за болезнь, чем опасна и как помочь ребенку.
Обновлено: 11.09.2024
Буллезный эпидермолиз - группа наследственных заболеваний, которые характеризуются легкой ранимостью кожи, отсюда второе название этих патологий - «механобуллезная болезнь». Основным симптомом служит развитие на поверхности кожных покровов пузырей с серозным содержимым, после чего на их месте возникают долго незаживающие эрозии. Диагностика различных типов буллезного эпидермолиза осуществляется при помощи иммуногистологических и генетических методик, а также на основании данных осмотра пациента и изучения его наследственного анамнеза. Специфического лечения не существует, однако правильная и комплексная симптоматическая терапия может в ряде случаев значительно улучшать состояние больного.
Лечение
Препараты (действующие вещества), применяющиеся при лечении
| Алюминия гидроксид (Aluminium hydroxide) |
| Амоксициллин (Amoxicillin) |
| Бацитрацин (Bacitracin) |
| Бетаметазон (Betamethasone) |
| Гентамицин (Gentamicin) |
| Гидрокортизон (Hydrocortisone) |
| Гидроксиметилхиноксалиндиоксид (Диоксидин) (Hydroxymethylquinoxalindioxide) |
| Дезлоратадин (Desloratadine) |
| Декспантенол (Dexpanthenol) |
| Ибупрофен (Ibuprofen) |
| Клавулановая кислота (Clavulanic acid) |
| Клиндамицин (Clindamycin) |
| Клобетазол (Clobetasol) |
| Клотримазол (Clotrimazole) |
| Левоцетиризин (Levocetirizine) |
| Лоратадин (Loratadine) |
| Магния гидроксид (Magnesium hydroxide) |
| Метилпреднизолон (Methylprednisolone) |
| Метронидазол (Metronidazole) |
| Моксифлоксацин (Moxifloxacin) |
| Мометазон (Mometasone) |
| Мупироцин (Mupirocin) |
| Натамицин (Natamycin) |
| Неомицин (Neomycin) |
| Нитрофурал (Nitrofural) |
| Парацетамол (Paracetamol) |
| Ретинол (Retinol) |
| Сульфатиазол серебра (Sulfathiazole silver) |
| Триамцинолон (Triamcinolone) |
| Фексофенадин (Fexofenadine) |
| Флуоцинолона ацетонид (Fluocinolone acetonide) |
| Хлоргексидин (Chlorhexidine) |
| Холина салицилат (Choline salicylate) |
| Цетиризин (Cetirizine) |
| Ципрофлоксацин (Ciprofloxacin) |
| Эбастин (Ebastine) |
Заключение
С тех пор как в 1886 году немецкий врач Кебнер впервые употребил термин «буллезный эпидермолиз», многое стало известно о патогенезе этого заболевания. Тем не менее до сих пор основным инструментом терапии является правильный уход за такими больными. Всё дело в том, что разработка и производство лекарств для лечения орфанных заболеваний представляет собой наукоемкий и дорогостоящий процесс, увы, невыгодный с коммерческой точки зрения из-за малого потребительского рынка. К сожалению, государство не в состоянии самостоятельно обеспечить решение этой проблемы. В 1978 году в Великобритании была основана ассоциация DEBRA, в настоящее время насчитывающая более 40 стран-участниц. Деятельность ассоциации направлена в первую очередь на медицинскую и социальную помощь пациентам с БЭ и их родным. Помимо этого, ассоциация финансирует исследования в области БЭ и занимается привлечением общественного внимания к данной проблеме. Представителем ассоциации в России является фонд «Б.Э.Л.А. Дети-бабочки». Именно благодаря совместным усилиям государства и ассоциации DEBRA стало возможным проведение масштабных исследований БЭ. В результате многолетней работы ученых всего мира появилось несколько перспективных подходов к терапии БЭ, а некоторые из них сейчас проходят первую фазу клинических испытаний. И хотя впереди еще несколько этапов тщательных исследований, первые положительные результаты дают надежду пациентам с этим редким генетическим заболеванием.

Жалобы:
· полушаровидные напряженные пузыри на коже туловища и конечностей, а также в ротовой полости;
· длительно заживающие эрозии;
· ранимость кожных покровов, чувствительность к любым механическим воздействиям;
· уплотнение, деформация или потеря ногтей пальцев рук и ног;
· облысение;
· зуд в области пузырей.
Анамнез:
· наследственность: наличие буллёзного эпидермолиза у родственников 1-й и 2-й степени родства;
· сезонность заболевания: при простом типе буллёзного эпидермолиза наблюдается четко выраженная сезонность заболевания – обострение и ухудшение состояния кожных покровов в летний период;
· аллергоанамнез при буллёзном эпидермолизе не отягощен;
· наличие сопутствующих заболеваний. При буллёзном эпидермолизе наблюдаются множественные внекожные проявления: микростомия, анкилоглоссия, аномалии эмали и дисплазия зубов, кариес, поражение органов зрения, патология ЛОР-органов, поражение ЖКТ и сердечно-сосудистой системы, деформации и контрактуры опорно-двигательной системы, поражение мочевыводящей системы.
Физикальное обследование:
· полушаровидные напряженные пузыри, наполненные прозрачной бесцветной жидкостью, на коже и слизистых оболочках в ротовой полости, в пищеводе и на верхних дыхательных путях, затрудняющие дыхание и питание пациента;
· длительно заживающие эрозии;
· ранимость кожных покровов, чувствительность к механическим и температурным воздействиям;
· ониходистрофия или отсутствие ногтевых пластинок;
· алопеция;
· ладонно-подошвенная кератодермия;
· милиумы;
· пигментные невусы.
Лабораторные исследования (УД – В)[3,7,8,9]:
· Гистологическое исследование биоптата кожи из очага поражения со свежим пузырем определяет субэпидермальную полость, но не позволяет диагностировать тип БЭ.
· Трансмиссионная электронная микроскопия: Простой БЭ – расслоение на уровне базального слоя эпидермиса, под ядрами клеток или выше, возможно скопление кератиновых филаментов. При пограничном БЭ – расслоение кожи на уровне светлой пластинки базальной мембраны, малые размеры и/или количество гемидесмосом. При дистрофическом БЭ – расслоение кожи непосредственно под плотной пластинкой базальной мембраны, малое количество или полное отсутствие крепящих фибрилл.
· Реакция непрямой иммунофлюоресценции (нРИФ) – выявляет экспрессию структурных белков кожи в месте дермоэпидермального сочленения (присутствие, снижение или отсутствие), уровень образования пузыря (внутриэпидермально, внутри светлой пластинки базальной мембраны, под плотной пластинкой базальной мембраны).
Инструментальные исследования: нет.
Диагностический алгоритм

Протеиновая терапия
При этом подходе в организм пациента вводится достаточное количество нормального белка. Исследования показали, что внутридермальное введение мышам с рецессивным дистрофическим БЭ очищенного человеческого коллагена VII приводит к формированию нормальных коллагеновых фибрилл в зоне соединения эпидермиса с дермой. При внутривенном введении очищенного белка наблюдается системное отложение коллагена VII в коже. Также было показано, что подобная терапия не только улучшает функциональное состояние кожи, но и способствует заживлению ран. Основываясь на результатах доклинических исследований, компания Lotus Tissue Repair, впоследствии вошедшая в состав Shire Pharmaceuticals, начала проведение расширенных доклинических исследований внутридермальных и внутривенных инъекций коллагена VII на модели рецессивного дистрофического БЭ у собак, а также первую стадию клинических исследований [3, 5].
Диагностика буллезного эпидермолиза
В настоящее время диагностика буллезного эпидермолиза осуществляется путем осмотра кожных покровов пациента, с помощью проведения иммуногистологических исследований и генетических анализов, в некоторых случаях производят изучение наследственного анамнеза. При осмотре кожных покровов специалист также может произвести диагностические тесты - механически воздействовать на кожу пациента и спустя время оценить результаты. Развитие на этом участке характерных для буллезного эпидермолиза пузырей или эрозий говорит в пользу наличия данного заболевания. На следующих этапах диагностики производят более точное определение формы патологии.
Иммунофлуоресцентный анализ при буллезном эпидермолизе осуществляется при помощи моно- и поликлональных антител, имеющих сродство к основным белкам эпидермиса, светлой пластинки и верхних слоев дермы. Это позволяет оценить количество того или иного белка, что, в свою очередь, говорит о ферментной активности тканей. Уменьшение количества того или иного белка свидетельствует о его низком выделении или же ускоренном разрушении. Снижение концентрации ключевых протеинов на определенных участках позволяет определить уровень развития пузырей на самом раннем этапе, что уже помогает с высокой долей вероятности определить тип буллезного эпидермолиза. Точку в диагностике этого состояния ставит генетический анализ методом прямого секвенирования генов, которые ассоциированы с тем или иным типом заболевания. Такой многостадийный подход к диагностике буллезного эпидермолиза обеспечивает высокую точность.
Значительно упростить диагностику этого заболевания позволяет изучение наследственного анамнеза пациента, по которому можно выявить его кровных родственников с такой же проблемой. Кроме того, если у кого-то из родных имеется буллезный эпидермолиз, имеет смысл производить пренатальную генетическую диагностику, что позволит выявить наличие данной патологии на ранних этапах развития плода. Дифференциальную диагностику осуществляют с истинной пузырчаткой, некоторыми формами буллезного пемфигоида, приобретенным буллезным эпидермолизом (который является не наследственным, а аутоиммунным заболеванием).
Генная терапия
Наконец, последний и, пожалуй, самый перспективный подход — генная терапия (рис. 6). В этом случае пациентам пересаживаются аутологичные (сделанные из собственных клеток) трансплантаты, в клетках которых с помощью методов генетического редактирования дефектный ген заменяется нормальным. И хотя подобная процедура не приводит к полному исцелению, она способна временно устранить симптомы БЭ, а также является относительно простой в техническом отношении.

Рисунок 6. Редактирование собственной ДНК в лечении БЭ. а — Принцип генной терапии. На первом этапе культивируют эпидермальные стволовые клетки из биоптатов кожи пациента с БЭ. Затем стволовые клетки подвергают процедуре генетического редактирования, в результате которой дефектный ген замещается нормальным. После того как пласт из генетически отредактированных кератиноцитов будет проверен на безопасность, его трансплантируют пациентам. б — Трансплантаты, сформировавшиеся из «правильных» кератиноцитов пациента с рецессивным дистрофическим БЭ. Рисунок из [7].
Первые успехи генной терапии были достигнуты в Италии в 2006 году, когда в эпидермальные стволовые клетки пациента с пограничным БЭ с помощью ретровирусной конструкции был доставлен нормальный ген LAMB3, кодирующий одну из субъединиц белка ламинина-332. Из «исправленных» клеток были созданы эпидермальные трансплантаты, которые затем были пересажены на ноги пациента. В результате из них образовалась полноценная в функциональном отношении кожа. Исследование показало, что выживание даже небольшого числа стволовых клеток в подобных трансплантатах приводит к успешному восстановлению нормальных функций кожи. Спустя восемь лет продукция целевого белка LAMB3 в коже пациентов всё еще сохранялась. Не было обнаружено ни пузырей, ни признаков воспаления, опухолевого роста или какого-либо иммунного ответа в области трансплантата. В июле 2014 года в Австрии провели вторую подобную операцию. Профессор Альфред Лэйн из Стэнфордского университета (Калифорния) вместе с сотрудниками начал клинические испытания подобной технологии, нацеленной на восстановление синтеза коллагена VII в эпидермальных стволовых клетках пациента с рецессивным дистрофическим БЭ. Спустя 30 дней после операции в областях трансплантации не было отмечено никаких отклонений, а уровень продукции коллагена VII был гораздо выше исходного [7].
Конечно, и в случае генной терапии существует множество сложностей. Так, замена гена с использованием вирусных конструкций и невирусных систем редактирования ДНК (ZFN, TALEN и CRISPR/Cas9)* связана с риском встраивания гена не в то место генома, которое являлось мишенью. Оценить последствия такого встраивания довольно сложно. Поэтому такой подход требует большой осторожности.
* — Методы ZFN, TALEN и CRISPR/Cas9 основаны на сайт-специфическом действии нуклеаз in vivo. Эти системы обычно состоят из двух модулей, один из которых распознает целевую олигонуклеотидную последовательность, а другой режет цепи ДНК: «А не замахнуться ли нам на. изменение генома?» [8], «CRISPR-системы: иммунизация прокариот» [9], «Мутагенная цепная реакция: редактирование геномов на грани фантастики» [10]. — Ред.
Другая проблема подобной терапии — возможный риск развития иммунного ответа на экспрессируемый новый белок. Особенно велик риск для тех пациентов, чьи клетки несли мутации, полностью исключающие синтез определенного белка. В то же время известно, что у некоторых пациентов с БЭ есть участки кожи, на которых не образуются пузыри и продуцируется повышенный уровень белка. Это явление названо обратным мозаицизмом, или естественной генной терапией, и может предотвращать развитие иммунной реакции на синтезируемый в результате генной терапии белок. Более того, такие «самоизлечившиеся» клетки можно использовать для клеточной терапии, и тогда потребность в генной коррекции исчезнет. Однако до сих пор попытки создать трансплантаты на основе таких клеток у больных с пограничным БЭ не увенчались успехом из-за малого числа подобных клеток в пересаживаемом материале [11].
МКБ-10
Q81 Буллезный эпидермолиз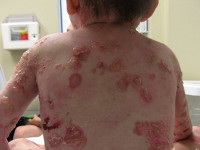
Классификация
В зависимости от уровня, на котором происходит повреждение кожи, выделяют четыре типа БЭ: простой, пограничный, дистрофический и синдром Киндлера.
Простой БЭ относится к так называемому интраэпидермальному типу БЭ, то есть повреждения затрагивают только эпидермис. Простой БЭ развивается в результате мутаций в генах белков, обеспечивающих прочность клеток базального слоя эпителия (рис. 2). Пузыри образуются ближе к поверхности кожи. Это самая легкая форма БЭ, при которой происходит полноценное заживление ран. Пограничный и дистрофический БЭ относятся к субэпидермальным типам БЭ, поскольку затрагивают не только эпидермис, но и дерму. При этих типах БЭ поврежденными оказываются белки, участвующие непосредственно в соединении двух слоев кожи (рис. 4). Полноценного заживления ран не происходит — на месте нормальной кожи в конечном счете образуются рубцы. Пограничный БЭ, в свою очередь, подразделяется на два типа: Герлиц и не-Герлиц, первый из которых летален. Дистрофический же БЭ в зависимости от типа наследования подразделяется на доминантную и рецессивную формы. Синдром Киндлера относится к смешанному типу БЭ, поскольку пузыри образуются на разных уровнях [1].

Рисунок 4. Иммунофлуоресцентное картирование образца кожи пациента с рецессивным дистрофическим БЭ. Для картирования использовали антитела к коллагену VII (зеленый цвет). а — Пониженное количество коллагена VII в биоптате от пациента с рецессивным дистрофическим БЭ. б — Кожа здорового донора. Ядра клеток окрашены йодидом пропидия (красный цвет). Рисунок из [3], с изменениями.
Дифференциальный диагноз
Симптомы буллезного эпидермолиза
Проявления буллезного эпидермолиза разных типов объединяет одно - развитие пузырей и эрозий в ответ на механическое воздействие на кожу. Различается лишь степень выраженности этих изменений, локализация, время существования и результаты заживления. При локализованной форме простого буллезного эпидермолиза (подтип Вебера-Коккейна) поражения располагаются только на определенном участке тела (руки, стопы). В младенческом возрасте возможна более широкая площадь появления пузырей, но с возрастом их выраженность уменьшается. Напротив, генерализованный подтип Доулинга-Меары характеризуется развитием мелких везикулярных высыпаний на значительной площади тела. Такой тип буллезного эпидермолиза возникает с самого раннего детства и может стать причиной смерти ребенка, итогом разрешения пузырьков может быть гиперкератоз, нарушения пигментации кожи, иногда возникает поражение слизистых.
Пограничная форма буллезного эпидермолиза протекает намного более тяжело, особенно так называемый летальный подтип Херлитца. При этом наблюдается повышенная ломкость кожных покровов, образование большого количества пузырьков, эрозий, на лице и спине часто возникают симметричные грануляции. Поражаются и слизистые оболочки рта, обнаруживается гипоплазия эмали и обусловленный ею тяжелый кариес. Столь тяжелое течение пограничного буллезного эпидермолиза часто становится причиной летального исхода в первые годы жизни. У выживших больных во взрослом возрасте формируются контрактуры суставов, поражение почек, потеря ногтей. Более легкая атрофическая форма пограничного буллезного эпидермолиза также характеризуется обширными высыпаниями, после разрешения которых формируются атрофические участки и рубцы. Также она часто приводит к дистрофии ногтей и рубцовой алопеции.
Дистрофический буллезный эпидермолиз практически всегда является генерализованным и поражает обширные участки тела. Доминантный вариант заболевания в целом отличается более доброкачественным течением, образование пузырей и их разрешение происходит медленно, однако большинство больных в конце концов теряют ногти на руках. После заживления эрозий на поверхности кожи формируются заметные рубцы. Рецессивный вариант дистрофического буллезного эпидермолиза, особенно его тяжелый генерализованный подтип, протекает намного тяжелее: помимо высыпаний у больных часто регистрируются псевдосиндактилии, обширные шрамы, потеря ногтей. Возникает поражение костей скелета, на месте заживших шрамов с годами может развиваться плоскоклеточный рак. Проблемой является еще и высокая устойчивость подтипа Аллопо-Сименса к терапевтическим мероприятиям.
Осложнения любого типа буллезного эпидермолиза сводятся к риску развития шока (при обширных поражениях), присоединения вторичной инфекции и спровоцированного ею сепсиса, обезвоживания больных. В большинстве случаев терапевтические процедуры производят только с целью недопущения этих состояний. Вероятность развития осложнений тем выше, чем большую область тела занимают патологические очаги и чем деструктивнее их характер (напряженные пузыри, эрозии, язвы).
Лечение буллезного эпидермолиза
Специфического лечения этого заболевания не существует, все терапевтические процедуры сводятся к предупреждению развития осложнений и уменьшению выраженности пузырьков и эрозий. В случае тяжелых форм буллезного эпидермолиза назначают преднизолон. Из наружных терапевтических манипуляций производят асептическое вскрытие пузырьков, обработку их крышки антисептиками, накладывают гелиомициновую мазь. Наложение повязок нужно производить крайне осторожно, так как давление бинтов может спровоцировать появление новых пузырей. При наличии осложнений (шока, сепсиса) проводят симптоматическое лечение противошоковыми препаратами и антибиотиками. С профилактической целью можно производить облучение кожных покровов ультрафиолетовыми лучами.
Современная генетика и ряд других областей медицины продолжают широкие исследования буллезного эпидермолиза с целью поиска более эффективных методик лечения. Среди основных технологий и методов наиболее перспективными считаются способы с использованием стволовых клеток, белковая и генная терапии. Однако пока ни один из методов не вышел за рамки экспериментов на животных, поэтому буллезный эпидермолиз в настоящее время является неизлечимым заболеванием.
Диагностика (стационар)
ДИАГНОСТИКА НА СТАЦИОНАРНОМ УРОВНЕ [3,14,15,16]
Диагностические критерии на стационарном уровне
Жалобы:
· полушаровидные напряженные пузыри, наполненные прозрачной бесцветной жидкостью на коже, а также в ротовой полости, в пищеводе и на верхних дыхательных путях, затрудняющие дыхание и питание пациента;
· длительно заживающие эрозии;
· ранимость кожных покровов, чувствительность к механическим воздействиям;
· уплотнение, деформация или потеря ногтей пальцев рук и ног;
· алопеция;
· зуд и/или болезненность в области пузырей;
· высыпания в полости рта и на половых органах;
· затруднение глотания пищи;
· деформации конечностей.
Анамнез:
· общим для всех форм буллёзного эпидермолиза является раннее время появления первых симптомов заболевания: возникновение пузырей и эрозий в местах малейшей механической травмы (давление и трение) кожи, при перепаде температуры окружающей среды, либо спонтанно сразу после рождения или первых дней (реже недель) жизни ребенка;
· наследственность: наличие буллёзного эпидермолиза у родственников 1-й и 2-й степени родства;
· сезонность заболевания: при простом типе буллёзного эпидермолиза наблюдается четко выраженная сезонность заболевания – обострение и ухудшение состояния кожных покровов в летний период;
· аллергоанамнез при буллёзном эпидермолизе не отягощен;
· наличие сопутствующих заболеваний. При буллёзном эпидермолизе наблюдаются множественные внекожные проявления: микростомия, анкилоглоссия, аномалии эмали и дисплазия зубов, кариес, поражение органов зрения, патология ЛОР-органов, поражение ЖКТ и сердечно-сосудистой системы, деформации и контрактуры опорно-двигательной системы, поражение мочевыводящей системы.
Физикальное обследование:
· полушаровидные напряженные пузыри, наполненные прозрачной бесцветной жидкостью, на коже и слизистых оболочках в ротовой полости, в пищеводе и на верхних дыхательных путях, затрудняющие дыхание и питание пациента;
· длительно заживающие эрозии;
· ранимость кожных покровов, чувствительность к механическим и температурным воздействиям;
· ониходистрофия или отсутствие ногтевых пластинок;
· алопеция;
· ладонно-подошвенная кератодермия;
· милиумы;
· пигментные невусы.
Лабораторные исследования: см. амбулаторный уровень.
Диагностический алгоритм
Перечень основных диагностических мероприятий:
· ОАК;
· ОАМ.
Перечень дополнительных диагностических мероприятий:
· патоморфологическое исследование биоптата кожи с последующей гистологией;
· электронно-микроскопическое исследование;
Общие сведения
Буллезный эпидермолиз - это гетерогенная группа наследственных заболеваний кожи, которые характеризуются образованием пузырей и эрозий в ответ на незначительное механическое воздействие. Впервые данный термин был использован в 1886 году немецким врачом-дерматологом Генрихом Кёбнером, дальнейшие исследования продемонстрировали, что существует множество разновидностей этой патологии. Генетические исследования буллезного эпидермолиза показали, что он может наследоваться как аутосомно-рецессивно, так и аутосомно-доминантно, с ним ассоциированы мутации более чем 10 генов. Существенные различия имеются и в клиническом течении разных типов этого заболевания, встречаемость колеблется в пределах 1:30000-1:1000000.
Патогенез нарушений при буллезном эпидермолизе долгое время оставался малоизученным. Прорыв в этом направлении произошел с внедрением в медицинскую практику электронной микроскопии, которая помогла визуализировать ультраструктуру пораженных тканей кожи. Следующий важный шаг в изучении буллезного эпидермолиза был совершен с открытием иммуногистологических исследований (иммунофлуоресценция). В настоящее время именно эти методики играют важнейшую роль в диагностике данных заболеваний, уступая по точности лишь генетическому анализу. Ввиду того, что методы изучения буллезного эпидермолиза постоянно совершенствовались, претерпевала изменения и классификация форм этой группы заболеваний.
Клеточная терапия
При этом способе лечения в организм пациента вводится необходимое и достаточное количество клеток, содержащих нормальный ген, кодирующий нужный белок.
Одним из перспективных видов клеточной терапии является внутридермальная инъекция аллогенных (полученных от здоровых доноров) фибробластов. Так, было показано, что подобный подход восстанавливает синтез коллагена VII и стабилизирует соединение эпидермиса с дермой в мышиной модели рецессивного дистрофического БЭ. Следующий шаг на пути к реализации этого вида терапии уже предпринят: нескольким пациентам с рецессивным дистрофическим БЭ были введены аллогенные фибробласты. И хотя спустя две недели после инъекции введенные клетки не обнаруживались, уровень продукции коллагена оставался повышенным как через две недели, так и через три месяца. Ученые считают, что основной терапевтический эффект инъекции аллогенных фибробластов заключается в увеличении продукции эпидермального фактора роста HB-EGF, а также в увеличении экспрессии гена коллагена VII в кератиноцитах и фибробластах реципиента. Однако механизм этого явления пока не установлен [3, 6].
Аллогенная трансплантация костного мозга также может быть перспективным способом лечения БЭ. Так, на мышиной модели рецессивного дистрофического БЭ было показано, что трансплантация костного мозга приводит к облегчению симптомов болезни. Вслед за обнадеживающими результатами были начаты клинические испытания подобного метода терапии. Однако пока результаты неутешительны: пять из двадцати пациентов умерли от прогрессии заболевания или осложнений после трансплантации [3, 6].
Прогноз буллезного эпидермолиза
Прогноз буллезного эпидермолиза чаще всего неопределенный, так как зависит от множества факторов и обстоятельств - типа заболевания, наличия или отсутствия у больного сопутствующих нарушений, его образа жизни. Например, локальный подтип простого эпидермолиза чаще всего имеет доброкачественное течение и редко создает угрозу жизни пациенту. Тогда как подтип Аллопо-Сименса имеет очень высокую смертность - как и от кожных проявлений, так и по причине отдаленных осложнений, таких как поражения почек и органов ЖКТ, а также развития плоскоклеточного рака кожи. Больные с такой проблемой должны бережно относиться к своей коже, не забывать про антисептическую обработку эрозий и других поражений, избегать занятий травмирующими видами спорта и иной деятельностью такого рода.

Автор
Редактор
Статья на конкурс «био/мол/текст»: «Мальчик, который остался без кожи». Это не название очередного фильма ужасов. Это документальный фильм режиссера Патрика Коллертона, запечатлевшего последние месяцы жизни 36-летнего Джонни Кеннеди, страдавшего редким генетическим заболеванием — дистрофическим буллезным эпидермолизом. Фильм привлек внимание пяти миллионов зрителей по всей Великобритании и помог собрать 500 000 фунтов стерлингов на благотворительность для международной ассоциации DEBRA, чья деятельность направлена на изучение и лечение буллезного эпидермолиза. Жизнь людей с этим диагнозом и их родных — это путь, который осилит не каждый: он тяжел как физически, так и морально, и требует мужества и невероятной воли к жизни. Детей, страдающих этим недугом, называют детьми-бабочками, сравнивая хрупкость их кожи с хрупкостью крыла бабочки. Что же такое буллезный эпидермолиз и каковы прогнозы на будущее?
Обратите внимание!
Эта работа опубликована в номинации «лучшая обзорная статья» конкурса «био/мол/текст»-2015.
Спонсором номинации «Лучшая статья о механизмах старения и долголетия» является фонд «Наука за продление жизни». Спонсором приза зрительских симпатий выступила фирма Helicon.
Спонсоры конкурса: Лаборатория биотехнологических исследований 3D Bioprinting Solutions и Студия научной графики, анимации и моделирования Visual Science.
Я знаю только один способ быть в ладу с собственной совестью: этот способ — не уклоняться от страдания.
Антуан де Сент-Экзюпери
Что такое буллезный эпидермолиз?
Буллезный эпидермолиз (БЭ) — группа редких генетически и клинически гетерогенных заболеваний, характеризующихся образованием пузырей и эрозий на коже и слизистых оболочках в результате незначительной механической травмы [1].
Кожа человека состоит из трех слоев: наружного — эпидермиса, среднего — дермы — и самого нижнего — подкожно-жировой клетчатки (гиподермы) (рис. 1).

Эпидермис кожи человека состоит из пяти различных слоев эпителиальных клеток, или кератиноцитов, самый нижний из которых — базальный эпителий — прикрепляется к дерме с помощью множества разных белков (рис. 2). Эти белки не только определяют стабильность соединения клеток базального эпителия с дермой, но и их собственную прочность. Мутации в генах, кодирующих эти белки, приводят к снижению прочности этого соединения, делая кожу чрезвычайно чувствительной к механическим воздействиям. Даже самое легкое надавливание — трение швов одежды, нежные объятия матери — может привести к образованию пузырей и причинить нестерпимую боль (рис. 3) [1].

Рисунок 2. Белки, участвующие в патогенезе различных типов наследственного БЭ. ПБЭ — простой БЭ; ПогрБЭ — пограничный БЭ; ДБЭ — дистрофический БЭ; СК — синдром Киндлера; БМ — базальная мембрана. Рисунок из [1], с изменениями.

Как правило, заболевание заметно сразу после рождения ребенка или в первые месяцы его жизни. Каждый новый день — настоящее испытание для больных БЭ и их опекунов: смена повязок, защищающих хрупкую кожу от повреждений и закрывающих уже существующие раны, обработка ран антисептиками — эти жизненно важные, чрезвычайно болезненные и изнурительные процедуры являются практически единственной составляющей современного лечения БЭ. При тяжелых формах заболевание поражает помимо кожных покровов слизистые оболочки и даже внутренние органы. БЭ относится к так называемым орфанным, то есть редким, заболеваниям. Частота встречаемости различных типов БЭ варьирует от 1:30 000 до 1:1 000 000 и зависит от популяции [2].
Лечение
К сожалению, в настоящее время БЭ неизлечим. Основная терапия направлена на предотвращение образования новых пузырей и эрозий, лечение ран и предотвращение их инфицирования. Главные задачи лечения — защита хрупкой кожи пациентов от механических воздействий (использование специальных повязок) и обработка уже существующих ран (применение антисептиков, наложение повязок).
Но наука не стоит на месте, и ученые всего мира отчаянно ищут лекарство от страшного недуга. Наиболее проработаны три перспективных вида терапии БЭ: протеиновая, клеточная и генная. Рассмотрим эти подходы подробнее.
Классификация буллезного эпидермолиза
В настоящий момент существуют десятки разновидностей буллезного эпидермолиза, которые достаточно трудно классифицировать в определенные группы. Проблема осложняется еще и тем, что почти за полтора века изучения данной патологии предпринимались неоднократные попытки разделить ее на определенные типы, используя самые современные на тот момент данные. В конечном итоге это привело к некоторой путанице, даже в научной литературе можно найти самые разнообразные варианты разделения буллезного эпидермолиза на разновидности. Наиболее современная классификация этого состояния в дерматологии включает в себя четыре типа заболевания, которые, в свою очередь, делятся на ряд подтипов:
- Простой буллезный эпидермолиз - имеет 12 подтипов, наиболее распространенными из которых являются синдромы Вебера-Коккейна, Кёбнера, Доулинга-Меары. Может наследоваться как аутосомно-доминантно, так и рецессивно, встречаемость составляет 1:100000. Простой буллезный эпидермолиз характеризуется образованием внутриэпидермальных или, реже, субэпидермальных пузырей, так как при этом заболевании поражаются белки эпидермиса.
- Пограничный буллезный эпидермолиз - делится на 2 подтипа, один из которых имеет еще 6 самостоятельных клинических форм. Наиболее тяжелой формой этого заболевания является подтип Херлитца, имеющий крайне высокую смертность. Встречаемость пограничного буллезного эпидермолиза составляет около 1:500000, образование пузырей при нем происходит на уровне светлой пластинки, что и дало ему название «пограничный».
- Дистрофический буллезный эпидермолиз - имеет два подтипа, которые делятся по механизму наследования этой патологии (доминантный и рецессивный подтипы). При этом встречаемость доминантного варианта несколько выше (3:1000000 против 1:500000 у рецессивной формы дистрофического буллезного эпидермолиза). Рецессивная разновидность также имеет несколько клинических форм, наиболее тяжелой из которых является подтип Аллопо-Сименса. При этом варианте заболевания у больных возникают глубокие эрозии, оставляющие после себя шрамы, возможны контрактуры суставов, поражение слизистых оболочек. Образование пузырей при этом происходит в сосочковом слое дермы, что и обуславливает появление шрамов и длительное заживление эрозий.
- Синдром Киндлера, или смешанный буллезный эпидермолиз, является одной из наиболее редких и малоизученных форм данной патологии. Особенностью, которая позволила выделить эту форму в отдельный тип, является образование пузырей во всех слоях кожи - эпидермисе, у светлой пластинке, в дерме. В настоящий момент определен только белок, выступающий в качестве мишени ферментов при смешанном буллезном эпидермолизе - киндлин-1.
Такой тип разделения всех клинических форм буллезного эпидермолиза является в настоящее время общепринятым. Но даже в пределах одного типа наблюдается большое разнообразие клинических симптомов заболевания, что осложняет диагностику и нередко влияет на прогноз патологии. Поэтому на сегодняшний день не прекращаются поиски более структурированной и приемлемой классификации буллезного эпидермолиза.
Лечение (амбулатория)
ЛЕЧЕНИЕ НА АМБУЛАТОРНОМ УРОВНЕ [3]
Тактика лечения [3, 14,15,16]: на амбулаторном уровне лечения буллёзного эпидермолиза предусмотрено проведение медикаментозной терапии поражений кожи и слизистых оболочек, уход за пораженной и непораженной кожей, а также немедикаментозное лечение (соблюдение режима, диетотерапия).
Немедикаментозное лечение
· Режим №2 (общий) – пациентам с БЭ рекомендуется избегать любых физических нагрузок, повышающих вероятность механического повреждения кожи и усиления потоотделения, резких движений и травмоопасных ситуаций. Перевязочные материалы, одежда и обувь пациентов должны быть изготовлены из натуральных тканей, без швов.
· Стол №15 (общий) – питание больных БЭ должно быть механически, термически и химически щадящим (протертое, полужидкое, негорячее), обогащенным основными макро- и микроэлементами, с высокой калорийностью. При многочисленности пузырей и эрозивных поверхностей пациент нуждается в восполнении теряемой жидкости.
· Уход за пораженной кожей. Напряженные пузыри прокалывают стерильной иглой с соблюдением правил асептики параллельно его покрышке, создавая 2 отверстия: входное и выходное. Содержимому пузыря дают вытечь, аккуратно помогая легкими промакивающими движениями. Не рекомендуется срезать покрышку пузыря во избежание обнажения раневой поверхности и провокации дополнительных болевых ощущений. Проколотые пузыри обрабатывают раствором антисептика, либо антисептическими или антимикробными кремами. Далее на раневые участки накладываются специальные перевязочные материалы для создания надежной защиты пораженных участков. Ежедневно сухие внешние слои повязок удаляют с помощью ножниц, а внутренние, прилипшие слои путем накладывания мокрых компрессов или отмачивания в ванной.
Медикаментозное лечение (УД – C, D) [15 –18]
Перечень основных лекарственных средств:
Антисептические средства для обработки вскрытых пузырей, эрозий и язв:
· хлоргексидина биглюконат 0,05%, 0,1%, 0,5% водный раствор;
· нитрофурал 0,02% раствор для местного и наружного применения (водный);
· гидроксиметилхиноксалиндиоксид 1% раствор для внутриполостного введения и наружного применения;
Стимуляторы регенерации тканей для эпителизации эрозий и язв:
· декспантенол + хлоргексидин, крем –1-2 раза в сутки наружно в течение 3 недель;
· декспантенол, крем – 1-2 раза в сутки наружно в течение 28 дней;
· ретинола ацетат в капсулах или в масляном растворе в суточной дозе 5000 МЕ на кг массы тела (равными частями утром и вечером после еды) перорально в течение 1,5–2 месяцев.
Перечень дополнительных лекарственных средств:
Топические антибактериальные и комбинированные средства при ограниченных инфицированных поражениях кожи:
· бацитрацин + неомицин, мазь – на инфицированные очаги поражения 2 раза в сутки в течение 10–17 дней;
· мупироцин, мазь 2% - на инфицированные очаги поражения кожи 3 раза в сутки в течение 7 дней;
· сульфатиазол серебра, крем - на инфицированные очаги поражения 2-3 раза в сутки в течение 10–14 дней, наружно или в виде окклюзионной повязки.
Алгоритм действий при неотложных ситуациях: нет.
Другие виды лечения:
· детям младшего возраста антирефлюксные смеси (УД – D) [21];
· гидротерапия курсом 10-15 процедур (УД – D) [22];
· В зависимости от типа БЭ, степени потребности в механической защите, объема экссудата, наличия бактериального обсеменения или вторичной инфекции подбирается перевязочный материал для покрытия раневой поверхности [13]:
a) для первичной перевязки:
- неадгезивные повязки;
- моделируемые повязки с мягким силиконовым покрытием.
b) при длительно существующих эрозиях и язвах (не менее 1 месяца):
- гидрогелевые, гидроактивные, гидроколлоидная повязка;
- коллагеновые пористые покрытия.
c) при выявлении признаков присоединения вторичной инфекции (гнойное отделяемое, гнойные корочки):
- серебросодержащие повязки;
- повязки, содержащие хлоргексидин;
- повязки, содержащие антибактериальный компонент.
Показания для консультации специалистов [3]:
· консультация генетика – для верификации диагноза и прогнозирования вероятности заболевания при повторных беременностях;
· консультация гастроэнтеролога – при наличии дисфагии и стриктур пищевода, рефлюкс-эзофагита, ЯБЖ и ДПК, хронической диареи/запоров, ректальных кровотечений, трещин заднего прохода и др.;
· консультация окулиста – при наличии пузырей и эрозий роговицы, рубцевания роговицы, эктропиона, обструкции слезных канальцев и т.д.);
· консультация оториноларинголога – при наличии отита среднего уха, стеноза наружного слухового прохода, тугоухости, утолщения и рубцевания голосовых складок, пузырей и изъязвления трахеи и гортани и др.;
· консультация стоматолога – при микростомии, анкилоглоссии, пузырях и эрозиях слизистой десен, аномалии эмали, дисплазии зубов, кариесе и др.;
· консультация хирурга – при наличии псевдосиндактилии, деформации по типу «варежки», контрактур, скрючевании дистальных фаланг, остеопорозе и остеопении;
· консультация уролога-нефролога – при наличии гидронефроза, гломерулонефрита, нефропатии, стриктур и дивертикулов мочеиспускательного канала, рубцевании головки полового члена, цистите, уросепсисе, почечной недостаточности и др.;
· консультация педиатра – при наличии пневмонии, анемии, иммунодефицитных состояний, снижении индекса массы тела и других состояний.
Профилактические мероприятия:
· медико-генетическая консультация;
· беседа с родителями о высоком риске рождения больного ребенка, а также о возможности мертворождения при высокой степени экспрессивности, а также летального исхода от сепсиса, пневмонии и др.;
· перинатальная диагностика;
· медико-социальная реабилитация;
· устранение факторов риска (механической травматизации, температурного и других видов воздействия);
· лечение сопутствующей патологии;
· использование лечебно-косметических средств;
· санаторно-курортное лечение.
Мониторинг состояния пациента:
· диспансерный учет по месту жительства у дерматолога, педиатра;
· наблюдение и лечение у смежных специалистов;
Индикаторы эффективности лечения:
· уменьшение площади поражения кожи и слизистых оболочек;
· регресс основных кожных высыпаний;
· отсутствие появления новых элементов;
улучшение общего состояния.
Диагностика
Для того чтобы подтвердить диагноз БЭ, необходимо провести тщательное обследование пациента. Самым важным этапом диагностики является исследование биоптата поврежденных участков кожи. Для этого используют такие методы, как световая, флуоресцентная и электронная микроскопия, а также генетический анализ.
Исследование с помощью световой микроскопии позволяет отличить интраэпидермальный тип БЭ от субэпидермальных. Для более точного определения уровня повреждения используют электронную и флуоресцентную микроскопии. С помощью электронной микроскопии можно отличить рецессивную форму дистрофического БЭ от доминантной (рис. 5). Так, при рецессивном дистрофическом БЭ наблюдается значительное снижение или полное отсутствие продукции коллагена VII типа, в то время как при доминантном дистрофическом БЭ дефектный белок вырабатывается в нормальном или слегка сниженном количестве. Однако из-за трудоемкости методов электронной микроскопии в настоящее время на первый план выходит микроскопия флуоресцентная. С помощью окрашивания флуоресцентными антителами этот метод позволяет визуализировать белки, участвующие в патогенезе заболевания, а также оценить их продукцию и распределение (рис. 4) [3].

Рисунок 5. Ультраструктура образца поврежденного участка кожи пациента с рецессивным дистрофическим БЭ. Звездочками обозначены места разрывов, расположенные сразу под темной пластинкой базальной мембраны (бесклеточного слоя, отделяющего дерму от эпидермиса), на которую указывают «галочки». Стрелками обозначены участки с единичными фибриллами коллагена VII. Изображение получено методом электронной микроскопии. Рисунок из [3].
Но только генетический анализ (ДНК-диагностику) можно назвать оптимальным методом для определения типа наследования и специфических мутаций, имеющихся у больных БЭ, а также наиболее точным методом различения клинических форм простого, пограничного и дистрофического БЭ [4].
Существует также пренатальная (до рождения) диагностика БЭ, материалом для которой служит ДНК из околоплодной жидкости, забираемая в первом триместре беременности (до 11 недель) [4].
Причины буллезного эпидермолиза
Этиология буллезного эпидермолиза неодинакова у разных типов заболевания, что в некоторых случаях достаточно сильно осложняет диагностику. Простой буллезный эпидермолиз обусловлен мутациями генов KRT5 и KRT14, однако, по данным врачей-генетиков, нарушением структуры этих генов объясняется только 75% случаев заболевания этого типа. При этом в кожных покровах, предположительно, нарушается равновесие в системе «ферменты-ингибиторы», и некоторые белки становятся объектом атаки. При простом буллезном эпидермолизе это могут быть протеины базальной мембраны (альфа6-бета4-интегрин) и белки десмосом базального слоя эпидермиса - десмоплакин, плакофиллин-1. В результате при механическом воздействии происходит выделение ферментов, которые разрушают указанные белки, тем самым провоцируя цитолиз и разрушение структуры эпидермиса, приводя к образованию пузырей.
Причиной развития другой формы патологии - пограничного буллезного эпидермолиза - являются мутации в генах LAMB3, LAMA3 и некоторых других. Большинство из этих мутации наследуется по аутосомно-рецессивному механизму, объектом атаки разбалансированной ферментной системы становятся такие протеины, как коллаген 17-го типа и ламинин-332. Эти белки участвуют в поддержании нормальной структуры нижних слоев эпидермиса, поэтому их повреждение приводит к характерным клиническим симптомам пограничного буллезного эпидермолиза. Помимо легкого образования пузырей и эрозий он характеризуется также повышенной ломкостью кожных покровов и более тяжелым течением.
Дистрофический тип буллезного эпидермолиза обусловлен мутациями в гене COL7A1, которые могут наследоваться как по аутосомно-доминантному, так и аутосомно-рецессивному механизмам. Белком-мишенью при этом выступает коллаген 7-го типа, который отвечает за стабильность структуры других соединительнотканных волокон кожи. Уменьшение количества этого протеина в тканях кожных покровов приводит к легкому развитию высыпаний, эрозий и пузырей, а также нередко сопровождается нарушениями других органов. В частности, дистрофический буллезный эпидермолиз часто приводит к развитию контрактуры суставов, поражение захватывает слизистые оболочки органов дыхательной и пищеварительной систем. На рубцах, которые остаются после заживления эрозий, нередко возникают злокачественные опухоли.
В целом, общий патогенез буллезного эпидермолиза можно свести к нарушению активности некоторых ферментов в тканях кожи. В результате этого разрушаются определенные ключевые структурные белки эпидермиса, дермы или базальной мембраны, что нарушает связи между клетками и приводит к образованию пузырей при механическом воздействии даже незначительной силы. Типы буллезного эпидермолиза отличаются один от другого локализацией пузырьков, видом мутации, что привела к этому заболеванию, и разновидностью белка, который стал объектом атаки ферментов.
Читайте также:
- Сицилийская борзая Чирнеко дель Этна: описание, фото, характер, история породы, особенности содержания и ухода
- Фото энтлебухер зенненхунда (74 фотографий)
- Выгодно или нет разведение овец как бизнес в домашних условиях для начинающих
- Английский кокер-спаниель (фото): Самый обаятельный охотник
- Кальфосет — инструкция по применению в ветеринарии