
Врожденное отсутствие соскового канала: клинические признаки, лечение
Обновлено: 18.09.2024
Появление на свет ребенка с врожденными дефектами развития всегда ошеломляет семью; эта тема - одна из самых тяжелых в акушерстве. Супруги в первый момент испытывают ни с чем не сравнимый психологический шок, который затем переходит в чувство вины, им кажется, что у них уже никогда не будет здорового ребенка.
Следует сразу сказать, что ребенок с врожденными пороками может появиться на свет абсолютно в любой семье - молодой, здоровой, без вредных привычек, с нормально протекающей беременностью. По данным многолетней статистики, во всем мире около 5% детей рождается с врожденными заболеваниями.
Врожденные пороки развития плода можно разделить на две большие группы - наследственно обусловленные (то есть заложенные в генах и хромосомах, передающиеся по наследству) и собственно врожденные (приобретенные в ходе внутриутробного развития). Такое деление довольно условно, так как большинство дефектов развития вызываются сочетанием наследственной предрасположенности и неблагоприятного внешнего воздействия, представляя собой мультифакториальные аномалии.
Проблема врожденных пороков развития плода очень многообразна, изучением этого вопроса занимаются различные специалисты - генетики, неонатологи, эмбриологи, специалисты по дородовой (пренатальной) диагностике. Разобраться в причинах всегда бывает непросто.
Наследственно обусловленные заболевания
В основе наследственно обусловленных заболеваний лежат мутации. Благодаря современным леденящим кровь триллерам, слово это вызывает сейчас у многих почти суеверный ужас. На самом деле латинское слово mutatio означает "изменение" - не более того. Мутация - это изменение наследственных свойств организма в результате перестроек в структурах, ответственных за хранение и передачу генетической информации. Заболевания, связанные с патологическими изменениями в хромосомах, обычно так и называют хромосомными заболеваниями. Под собственно наследственными заболеваниями понимают нарушения, обусловленные генными мутациями.
В ядрах соматических (неполовых) клеток содержится по 23 пары хромосом, из которых одна пара - половые хромосомы. У женщин эта пара состоит из двух одинаковых хромосом, условно называемых Х-хромосомами, у мужчин эти хромосомы разные - Х-хромосома и Y-хромосома. Неполовые хромосомы называются аутосомами.
В половых клетках хромосом в два раза меньше - не 23 пары, а 23 штуки.
При оплодотворении ядра яйцеклетки и сперматозоида сливаются, и будущий человечек получает полный набор хромосом, наследуя таким образом и материнские, и отцовские признаки.
Хромосомы состоят из генов. За каждый признак в организме отвечает пара генов - "мамин" и "папин". (Исключение составляет XY-пара половых хромосом у мужчин: не все гены Х-хромосомы имеют "напарников" в Y-хромосоме.) В каждой паре один ген доминирует (доминантный ген), т.е. проявляется обусловленный им вариант признака, другой - "уступает" (рецессивный ген). При неблагоприятном стечении обстоятельств оба гена в паре или один из них могут оказаться носителями патологического признака. В первом случае их "владелец", без сомнения, болен. Если же мы имеем дело лишь с одним "больным" геном, возможны два варианта: (1) за заболевание "отвечает" доминантный ген - тогда его носитель болен; (2) носитель патологического признака - рецессивный ген - тогда человек здоров (точнее, как говорят врачи, фенотипически здоров, т.е. при наличии "больного" гена в генотипе, отсутствуют какие-либо проявления болезни).
Результаты
При УЗ-исследовании обнаружен один живой плод женского пола в головном предлежании. Гестационный срок составил 34,4 нед. Фетометрические показатели плода соответствовали данному сроку. Патологических изменений плаценты и околоплодных вод не выявлено. При исследовании мочевой пузырь в типичном месте не визуализировался.
В нижних отделах передней брюшной стенки в надлобковой области визуализировалось эхогенное образование размером 30x28x25 мм (рис. 1, 2). Определялось низкое впадение пуповины. Почки плода визуализировались в типичном месте, без структурных изменений. Патологических изменений других органов не было выявлено.
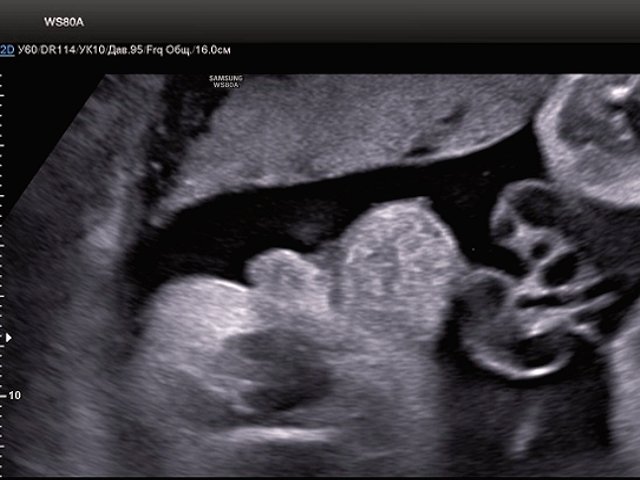
Рис. 1. Эхогенное образование в нижнем отделе передней брюшной стенки (экстрофированный мочевой пузырь).
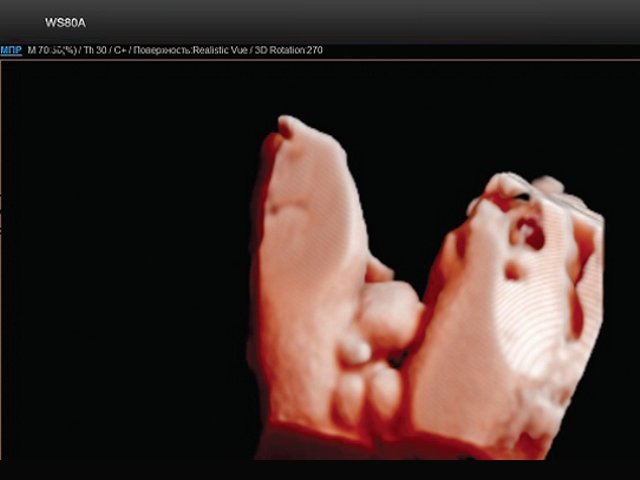
Рис. 2. Экстрофия мочевого пузыря в сочетании с отсутствием видимой патологии наружных половых органов (режим трехмерной реконструкции).
Поставлен диагноз: "Беременность 34,4 нед. ВПР плода - экстрофия мочевого пузыря".
Пренатальными УЗ-признаками экстрофии мочевого пузыря являются: отсутствие визуализации мочевого пузыря при нормальном количестве околоплодных вод, структурно неизмененные почки, визуализация эхогенного образования в надлобковой области, аномалии строения половых органов. Использование ЦДК является дополнительным инструментом для диагностики экстрофии мочевого пузыря. В норме пупочные артерии проходят вдоль стенки мочевого пузыря при поперечном сканировании нижней части живота. Для экстрофии мочевого пузыря характерно низкое впадение пуповины, при котором артерии охватывают образование в надлобковой области. При отсутствии визуализации мочевого пузыря в типичном месте необходимо провести повторное сканирование через 30-40 мин для адекватной оценки его расположения и степени наполнения.
Изолированная экстрофия мочевого пузыря, как правило, не сочетается с хромосомными перестройками у плода и не требует пренатального кариотипирования.
В данном клиническом наблюдении экстрофия мочевого пузыря была диагностирована у плода женского пола, что встречается значительно реже, чем у плодов мужского пола. Кроме того, отсутствие выраженной деформации наружных половых органов позволяет предположить неполную форму экстрофии. Оценка анатомии мочевого пузыря включена в протокол обязательного скринингового обследования во всех триместрах беременности. Отсутствие визуализации мочевого пузыря в типичном месте должно насторожить врачей УЗ-диагностики. Однако часто экстрофия мочевого пузыря диагностируют лишь в III триместре беременности.
После рождения детям с экстрофией мочевого пузыря требуется многоэтапная сложная хирургическая коррекция. Процент неудовлетворительных отдаленных результатов, к сожалению, остается на сегодняшний день очень высоким, как правило, вследствие присоединения таких осложнений, как нарушение функции тазовых органов: недержание мочи, рубцовая деформация половых органов, половая дисфункция, выраженная деформация передней брюшной стенки, уретерогидронефроз III-IV степени, непрерывно рецидивирующий пиелонефрит со снижением функции почек, хроническая почечная недостаточность и др. [4, 7]. Огромной и сложной проблемой остается медико-социальная реабилитация больных с экстрофией мочевого пузыря.
Литература
УЗИ сканер RS80
Эталон новых стандартов! Беспрецедентная четкость, разрешение, сверхбыстрая обработка данных, а также исчерпывающий набор современных ультразвуковых технологий для решения самых сложных задач диагностики.
По мнению директора по развитию специализированного центра восстановления зрения ОПТИМЕД врача Александра Нелина, очень важно следить за здоровьем глаз с рождения. Поэтому очень важно знать о проблемах и патологиях органов зрения у детей и о методах их лечения.
Врач Александр Нелин ознакомился со статьей «Непроходимость носослезного канала (NLDO) у детей», написанной авторами Shani Golan and Gary J Lelli Jr, и рассказывает об этом исследовании:
«Непроходимость носослезного канала (Nasolacrimal duct obstruction - NLDO) распространена в педиатрии и проявляется в виде стойкой эпифоры, рецидивирующего конъюнктивита, образования корок на веках и иногда в виде дакриоцистита. Она обычно врожденная и встречается на уровне клапана Хаснера. Варианты лечения педиатрической NLDO включают нехирургические и хирургические процедуры. Лечение может проводиться как на рабочем месте, так и в операционной. В статье «Непроходимость носослезного канала (NLDO) у детей» авторы обсуждают патогенез педиатрической NLDO и предоставляют обновленную информацию о существующих вариантах лечения, включая массаж, который остается очень успешным методом, и хирургическое вмешательство, которое может быть оправдано у детей в возрасте старше трех лет, у которых имеются анатомические вариации и черепно-лицевые аномалии, или у пациентов, не реагирующих на медикаментозную терапию и зондирование, а также у пациентов с приобретенным NLDO. Кроме того, авторы изучили преимущества новых технологий и эндоскопических подходов, в том числе с более коротким временем оперативного вмешательства и с отсутствием рубцов, а также возможность выполнять двусторонние процедуры и одновременно решать любую дополнительную интраназальную патологию».
Вот что пишут авторы в своей статье: «Непроходимость носослезного канала (NLDO) распространена в педиатрии. Анатомическое расположение непроходимости быть как до, так и после слезного мешка. Она обычно врожденная и возникает из-за сохранения мембраны на уровне клапана Хаснера в дистальном канале носоглотки. Другие причины включают в себя посттравматическую непроходимость слезной системы, наличие связанных системных аномалий и приобретенные причины NLDO. Заболеваемость выше у детей с черепно-лицевыми аномалиями.
Клиническое проявление детской NLDO обычно в виде эпифоры (которая поражает до 20% детей), рецидивирующего конъюнктивита и корки на веках. Время от времени возникает отек ниже уровня медиального кантуса (слезное мукоцеле). Если мукоцеле становится инфицированным, то его сопровождают припухлость и эритема. Если дакриоцистит принимает серьезную форму, может произойти разрыв зараженного мешка через кожу, что может привести к слезной фистуле. Если надавить на слезный мешок, может быть рефлюкс слизистого материала из пункции.
В этой обзорной статье мы (авторы) обсудим анатомию носослезной системы, патогенез детской NLDO и предоставим обновленную информацию о текущих вариантах лечения.
Анатомия носослезной системы
Слезы проходят через верхние и нижние слезные точки, через нижний и верхний канальцы к общему канальцу, который стекает в слезный мешок. В месте соединения между общим канальцем и слезным мешком находится клапан Розенмюллера (valve of Rosenmuller ). Клапан Розенмюллера представляет из себя односторонний клапан. Жидкость из слезного мешка стекает ниже в носослезный проток и оттуда в нижнее отверстие носа, которое частично покрыто слизистой оболочкой, известной как клапан Хаснера.
Эпифора в педиатрии
Врожденная непроходимость носослезного канала (Congenital nasolacrimal duct obstruction - cNLDO) является наиболее распространенной причиной педиатрической эпифоры и обсуждается в этом обзоре. Киста слезного мешка (Dacryocele) - еще один распространенный механизм и обсуждается позже. Другие причины педиатрической эпифоры обнаруживаются примерно в 4% случаев и включают проксимальный слезный дисгенез, точечный агенезис и дисгенезия канальцевой стенки.
В нескольких случаях пункционного и канального дисгенеза было отмечено, что средний возраст пациентов был старше, чем у пациентов с cNLDO (3,6-6,8 лет). Эти отличающиеся от cNLDO диагнозы должны быть тщательно оценены, прежде чем ставить диагноз cNLDO, поскольку требуемое лечение отличается.
Врожденная непроходимость носослезного канала
Врожденная непроходимость носослезного канала (Congenital nasolacrimal duct obstruction - cNLDO) встречается примерно у 6-20% новорожденных. Часто в этих случаях непроходимость проходит сама или с медицинской помощью в течение первого года жизни; однако более сложные случаи требуют хирургического вмешательства. В последние годы было опубликовано несколько исследований cNLDO, проведенных Группой по исследованию детских глазных болезней (Pediatric Eye Disease Investigator Group - PEDIG), включая исследования, посвященные связи между cNLDO и аметропией / амблиопией, а также посвященных представлению новых хирургических методов и инструментов для лечения этого состояние.
Развитие слезной системы начинается с пятой недели беременности. Просвет формируется в слезном канале, а кавитация слоя нижнего прохода происходит на 10-ой неделе. В дальнейшем, канализация слезного канала позволяет связаться с нижним проходом с шестого месяца плода до конца беременности. Если этого не происходит, на клапане Хаснера образуется мембранный барьер, который самопроизвольно разрешается в 85-95% случаев к первому году жизни.
Недавнее исследование с использованием компьютерной томографии с высоким разрешением показало, что препятствием является либо сохранение мембраны на дистальном конце канала, либо костная обструкция, либо сужение нижнего прохода с привязкой к слизистой оболочке носа.
Киста слезного мешка (Dacryocele)
Киста слезного мешка встречается примерно у одного из 3 900 новорожденных. В дополнении к типичной непроходимости на клапане Хаснера эти новорожденные имеют эффект шарового клапана на уровне клапана Розенмюллера, что приводит к разрыву слезного мешка. Киста слезного мешка проявляется как сильный отек на нижнем веке чуть ниже медиального кантуса и может быть обнаружена пренатально на ультразвуковом и магнитно-резонансном изображении. Это чаще всего встречается у женщин и является двусторонним у 25% пациентов. Инфекция возникает в 24-60% случаев.
Как и в случае с cNLDO, киста слезного мешка имеет высокую вероятность спонтанного разрешения; 50% случаев кисты слезного мешка разрешаются пренатально до рождения. Массаж слезного мешка и введение местных антибиотиков может привести к разрешению в 76% случаев. Эндоскопическое удаление связанной интраназальной кисты увеличивает вероятность успеха до 95%.
Лечение непроходимости носослезного канала (NLDO) у детей
NLDO имеет высокий уровень разрешения без хирургического вмешательства. В одном обзорном исследовании, использующем только медицинское лечение, уровень разрешения NLDO к первому году жизни был 80% в возрасте 3 месяцев, 70% в возрасте 6 месяцев и 52% в возрасте 9 месяцев. PEDIG обнаружил, что у 66% младенцев в возрасте 6-10 месяцев проблема разрешались без хирургического лечения в течение 6-месячного периода. Также возможно спонтанное разрешение в возрасте старше 1 года, о чем свидетельствуют исследования Yound et al., которые обнаружили, что у 41% детей с врожденным NLDO происходило спонтанное разрешение во второй год жизни (после 1 года, но до достижения 2 лет ).
Медицинское лечение
Медицинское лечение NLDO состоит из массажа носослезного мешка вместе с использованием местных антибиотиков, если имеются выделения. Массирование носослезного мешка нисходящими движениями приведет к гидростатическому давлению, нарушающему мембранную обструкцию на клапане Хаснера.
Нисходящий массаж носослезного мешка более эффективен, чем простой массаж или отсутствие массажа. Недавнее исследование показало, что успешность техники массажа Криглера, составляла 56 % у детей в возрасте до 2 месяцев, 46% у детей в возрасте 2-6 месяцев и у 28% у детей старше 6 месяцев.
Зондирование носослезного канала
В целом, показатель успешности зондирования оценивается в диапазоне 80-90% у детей в возрасте до 3 лет и остается высоким до достижения ребенком трехлетнего возраста, после чего показатели успеха начинают снижаться. Исследования, проведенные PEDIG, у детей в возрасте до 4 лет с cNLDO показали, что показатель успеха зондирования составляет 78%, 82%, с баллонным катетером, и 91% с интубацией носослезного канала. Показатель успеха ниже при двустороннем заболевании или когда присутствует более одного клинического признака NLDO, но, по-видимому, он не связан с возрастом в пределах от 6 до <15 месяцев.
С внедрением эндоскопических методов хирургических вмешательств при операциях на носослезной системе, эндоскопическое зондирование было предложено в качестве альтернативы традиционному слепому зондированию. Эндоскопическое зондирование позволяет визуализировать нижнюю часть носослезной системы и носовые аномалии, такие как атрезия, киста, стеноз и ложный проход для зондирования. Кроме того, прямая визуализация позволяет наблюдать сам процесс прохождения зонда, что, в свою очередь, влияет на успех зондирования. Несколько исследований, сравнивающих эти два метода у детей с cNLDO, показали значительно более высокий уровень успеха при эндоскопическом зондировании, особенно у детей старшего возраста, вероятное это связано с дополнительными возможностями наблюдать и лечить связанные проблемы.
Интубация силиконовой трубки для лечения cNLDO является хорошо известным вариантом лечения cNLDO после неудачного зондирования и орошения. В нескольких крупных исследованиях сообщается о показателях успеха в диапазоне от 82-97% после силиконовой интубации. Противоречие существует в отношении того, может ли назолакримальная интубация с большей вероятностью потерпеть неудачу у детей старшего возраста. В сравнительном исследовании простого зондирования и зондирования с интубацией в качестве вторичной процедуры, у пациентов, получавших интубацию, значительно чаще наблюдался успешный результат.
Биканаликулярная и моноканаликулярная интубация одинаково успешны при лечении cNLDO. Моноканаликулярную трубку легче вставлять и удалять, и поэтому она чаще используется в педиатрии. Большинство интубационных систем требуют интраоперационного извлечения трубки из носа. Однако моноканаликулярные трубки могут быть удалены проксимально, что позволяет проводить удаление в офисе у многих детей. Лакримальные трубки обычно удаляются через 2-6 месяцев. У детей в возрасте до 2 лет удаление трубки обычно происходит через 6 недель, для детей старшего возраста подходит более длительный период интубации, и шансы на успех возрастают, если трубка остается на месте не менее 3 месяцев.
Несколько состояний могут потребовать более раннего удаления трубки, например абразия (травма) роговицы или конъюнктивы от трубки, образование гранулем и пункционная эрозия.
Моноканаликулярные трубки Masterak® (FCI Ophthalmics Inc., Pembroke, MA, US) удерживаются на месте с помощью крючка на опорной пластине, который помещается в слезную точку. Эта опорная пластина может расшататься или самопроизвольно выпасть, поэтому следует проявлять осторожность при расширении слезной точки, чтобы не увеличить ее до размеров, когда они не сможет надежно удерживать опорную пластину трубки. Биканаликулярные трубки, если они размещены в носу, могут потребовать удаления под кратковременной общей анестезией.
Педиатрическая дакриоцисториностомия (DCR)
Дакриоцисториностомия (Dacryocystorhinostomy -DCR) у детей применяется в случаях NLDO, которые не реагируют на медикаментозную терапию, зондирование или интубацию. Когда спрашивают о сроках хирургического вмешательства, 79% членов Американской ассоциации детской офтальмологии и косоглазия ответили, что они начинают рекомендовать хирургическое вмешательство под общей анестезией для случаев неразрешенного cNLDO в возрасте с 13 месяцев.
Несмотря на техническую сложность проведения у маленьких детей, эндоназальная DCR обладает несколькими преимуществами по сравнению с внешней: она может быть выполнена в острых состояниях, она позволяет избежать рубцов на лице, она вызывает меньше анатомических нарушений, она не нарушает механизм слезного насоса, она уменьшает время оперативного вмешательства, и она может даже уменьшить послеоперационный дискомфорт. Дополнительно, с помощью эндоназальной DCR можно одновременно лечить любую связанную носовую патологию и она может выполняться с двух сторон под общей анестезией в качестве амбулаторной процедуры. Однако недостатки, связанные с эндоназальной DCR, включают более длительное обучение работе в узком носовом пространстве у детей и технические трудности, возникающие из-за существующих анатомических разнообразий. При сравнении успешности эндоназальной и внешней DCR в педиатрии, эндоскопическая DCR дает аналогичные результаты по сравнению с внешней.
В 2011 году Uysal et al. сообщили о своем опыте проведение процедуры с использованием эндоканалического диодного лазера. Из представленных 18 случаев, в 100% достигнут анатомический успех (определяемый проходимостью эндотелия); клинический показатель успеха (разрешение эпифоры) составил 85%.
Кроме того, ультразвуковая эндоскопическая DCR является альтернативным методом управления NLDO. Основным преимуществом этого метода является безопасность для окружающих мягких тканей во время остеотомии. В недавнем исследовании, опубликованном на 6-м месяце наблюдения, анатомический и функциональный успехи были отмечены у 93,1% и 88,6% соответственно.
В нашем обзоре рассматриваются клинические аспекты детской NLDO. Массаж с использованием техники Криглера (Crigler maneuver) остается весьма успешным. Зондирование слезной системы также очень эффективно, по крайней мере, до 3-х летнего возраста. Разрешение с помощью медицинского вмешательства возможно после первого года жизни, поэтому разумно продолжать до примерно 15-месячного возраста. После неудачных попыток зондирования следует применять интубацию носослезного канала. Кроме того, дети с NLDO должны наблюдаться до 3-4 лет, чтобы убедиться, что не развивается анизометропическая амблиопия.
В некоторых случаях может потребоваться операция DCR, эти случаи включают: дети старше 3 лет, дети с с анатомическими вариациями и черепно-лицевыми аномалиями, пациенты, не отвечающие на медицинскую терапию и зондирование, и пациенты с приобретенной NLDO. Хотя внешняя DCR остается успешным и жизнеспособным вариантом, новые технологии и эндоскопические подходы позволяют сократить время оперативного вмешательства и не оставляют рубцов. Кроме того, эти подходы позволяют выполнять двусторонние процедуры и позволяют врачу одновременно решать любую дополнительную интраназальную патологию.

Профессиональные диагностические инструменты. Оценка эластичности тканей, расширенные возможности 3D/4D/5D сканирования, классификатор BI-RADS, опции для экспертных кардиологических исследований.
Настоящая работа посвящена изучению плодового фенотипа при ультразвуковом исследовании в различные сроки беременности при помощи новейших технологий Samsung, оптимизирующих диагностику многих врожденных пороков развития, аномалий, нарушений строения, особенностей развития с оценкой значимости отдельных ультразвуковых патологических маркеров в качестве потенциальных признаков генетических синдромов и ассоциаций.
Одной из ведущих причин детской и младенческой смертности являются врожденные пороки развития (ВПР), среди которых в 20% случаев встречаются множественные ВПР (МВПР) с установленной частотой 1:250 новорожденных [1]. Особое значение в профилактике МВПР имеет пренатальная диагностика - современный и высокоэффективный метод диспансеризации плода, направленный на своевременное выявление патологии, впоследствии определяющий выбор адекватной акушерской тактики при данной беременности и специфических мер профилактики болезни в данной семье в дальнейшем [2]. Современные ее возможности позволяют из комплекса патологических нарушений у плода выделить достоверно значимые ультразвуковые маркеры для постановки диагноза генетических синдромов различной этиологии. Сегодня пренатальная синдромология активно развивается, как в плане расширения спектра возможных лабораторных исследований, так и описания значимых, ключевых, таргетных ультразвуковых признаков многих наследственных синдромов различного происхождения.
Группа МВПР включает сотни нозологических форм, различных по этиологии, фенотипическим проявлениям и прогнозу. В силу огромной гетерогенности МВПР представляют собой сложную проблему для постановки окончательного диагноза. Основными этиологическими факторами, приводящими к возникновению синдромов с множественными пороками развития, являются генные, хромосомные мутации и действие на плод неблагоприятных факторов внешней среды [3, 4].
Любые наследственные или врожденные генетические болезни являются результатом повреждения генетической информации на геномном, хромосомном или генном уровне. В 60% всех случаев МВПР встречаются хромосомные синдромы, достаточно широко изученные и хорошо диагностируемые. Для лабораторной диагностики этих повреждений разработаны разнообразные и хорошо известные методы: цитогенетический, молекулярно-цитогенетический и молекулярно-генетический. МВПР нехромосомного генеза встречаются в 40% случаев. Среди них выделяют синдромы, ассоциации и неклассифицированные комплексы. Применительно к нехромосомным синдромам цитогенетическое исследование позволяет лишь отвергнуть хромосомную природу данного комплекса пороков развития, но не установить диагноз.
Сегодня имеется определенное количество публикаций о выявлении некоторых синдромов и ассоциаций при пренатальной эхографии [6, 7]. Однако дородовая идентификация нозологических форм нехромосомных синдромов пока не вошла в рутинную практику врачей в области пренатальной диагностики и носит случайный характер. В литературе отсутствуют четкие рекомендации о системном подходе к диагностике наследственных синдромов различной этиологии, нет диагностических алгоритмов и определенных схем взаимодействия врачей ультразвуковой пренатальной диагностики, генетиков, акушеров-гинекологов.
Использование эхографии делает возможным пренатальное выявление не менее 80% врожденных дефектов во II, III триместрах беременности и свыше 50% значимых нарушений анатомии плода уже в I триместре начиная со сроков 11-12 нед. Весь комплекс диагностированных пороков развития и различных микроаномалий в совокупности составляет фенотипический спектр синдрома, который состоит из «ядерных» (главных, ключевых) признаков и дополнительных аномалий, наличие которых не является обязательным для диагностики клинической нозологии синдрома.
Революцией в пренатальной ультразвуковой диагностике явилось появление объемной эхографии, которая, обладая такими качествами, как неинвазивность, безопасность и возможность многократного применения у одной пациентки, имеет высокую информативность в исследовании анатомии плода и изучении его фенотипа. При применении различных режимов объемной эхографии абсолютно очевидно их преимущество по сравнению с обычным сканированием. Детально можно изучить лицо плода (рис. 1-4) в различные сроки беременности, начиная со сроков первого пренатального скрининга в 11-14 нед, конечности плода, причем не только их наличие и положение (рис. 5, 6), но и состояние и количество пальцев (рис. 7-9) как на руках, так и на ногах. Также можно изучить позвонки плода (рис. 10), состояние твердого нёба (рис. 11, 12), строение наружного уха (ушной раковины) (рис. 13), состояние основных швов черепа и родничков, исключая их преждевременное закрытие при кранисиностозах (рис. 14, 15).
Манифестные признаки синдрома ЕЕС (синдромальное ядро)
Эктродактилия - это расщелина кистей и/или стоп с отсутствием одного или нескольких центральных пальцев, известная также под названием "Split hand-split foot malformation" (SHFM). Часто внешний вид кистей и стоп сравнивают с клешней краба. Нередко встречается в сочетании с синдактилией (сращением пальцев).
Эктодермальная дисплазия проявляется особенностями строения всех производных эктодермы. Изменения затрагивают волосы, зубы, ногти [11]. Волосы и ресницы у таких людей тусклые, редкие и жесткие. Характерны аномалии зубов, гипоплазия зубной эмали, снижение пигментации волос, кожи, радужной оболочки глаз, что вызывает светлый их цвет. Также часто сочетание синдрома с фотобоязнью [1], обструкцией носо-слезных каналов [12], кондуктив- ной тугоухостью [13].
Эктодермальная дисплазия проявляется отсутствием потовых желез, что характеризуется наличием гипогидротинового синдрома, проявляющегося в резкой сухости и шелушении кожи. Характерным проявлением этого синдрома является хриплый, осипший голос из-за нарушения увлажнения голосовых связок, вследствие чего не происходит полного их смыкания [14].
Нарушение речи у больных с ЕЕС-синдромом имеет много причин. Во-первых, это связано с последствиями наличия расщелины губы и/или неба и "гиперназальной" речью, во-вторых, правильной артикуляции может препятствовать не только изменение голосовых связок, но и патология зубов. В-третьих, из-за аномалий слуховых косточек часто прогрессирует тугоухость, ведущая к когнитивным расстройствам и расстройствам речи.
Дети с этим синдромом обычно имеют нормальный уровень интеллекта. Лечение основано на коррекции пороков лица и конечностей. Признаки эктодермальной дисплазии проявляются в постнатальном периоде [1]. Врожденные пороки развития (ВПР) мочевыделительной системы у больных с синдромом ЕЕС возникают более чем в половине случаев. Среди них описаны: мегауретер, гидронефроз, уретероцеле, аплазия почки, аномалии гениталий, уретровезикальный рефлюкс с частыми инфекциями мочевыделительных путей [12].
Пренатальная диагностика синдрома ЕЕС
Учитывая выраженность проявлений при синдроме ЕЕС, пренатальная диагностика этой патологии, безусловно, возможна. Однако на сегодняшний день в мировой литературе встречается незначительное число публикаций, посвященных диагностике этого редкого синдрома 15, что связано, видимо, с редкостью возникновения данной аномалии. В основном сроками постановки диагноза являются 16-30 недель беременности. Большую помощь в диагностике данной патологии, учитывая выраженные изменения фенотипа лица и конечностей, оказывает применение новых ультразвуковых технологий 3D/4D с методиками поверхностной реконструкции [16].
Первое описание пренатальной диагностики синдрома датировано 1993 годом и принадлежит M. Bronshtein и R. Gershoni, когда при применении трансвагинальной эхографии у плода была выявлена расщелина губы и неба и эктродактилия в 14 недель беременности [17]. Уникальность этого факта в том, что, пожалуй, впервые из известных случаев диагностики врожденных аномалий дебют дородового выявления порока принадлежит сроку первого триместра. Примечательно и то, что авторами не только описаны отдельно выявленные пороки развития плода, но и пренатально установлена нозология этого состояния. Другими словами, диагноз не звучал, как "множественные врожденные пороки развития", когда невозможно говорить об этиологии заболевания, а следовательно, и о мерах специфической профилактики данной патологии в дальнейшем в семье. Пренатальный диагноз был выставлен полно и абсолютно корректно, что особенно важно при проведении медико-генетического консультирования с формированием тактики адекватного репродуктивного поведения семьи в дальнейшем.
Из-за незначительного количества публикаций, посвященных пренатальной диагностике этого генетического синдрома, представляем ряд собственных наблюдений диагностики синдрома ЕЕС в разные сроки беременности, в том числе и в первом триместре. Особо рассмотрим особенности проведения медико-генетического консультирования при диагностике различных форм синдрома с аутосомно-доминантным типом наследования для выработки специфических мер профилактики данного наследственного заболевания.
Врожденные мультифакториальные пороки развития
- Ионизирующее излучение (рентгеновские лучи, воздействие радиоактивных изотопов). Кроме прямого действия на генетический аппарат, ионизирующее излучение обладает токсическим эффектом и является причиной многих врожденных аномалий
- Тератогенные инфекции, т.е. инфекционные заболевания, передающиеся от матери плоду
- Медикаменты. Нет лекарств, которые могут быть безоговорочно признаны полностью безопасными, особенно на ранних стадиях беременности. Во время беременности лучше воздержаться от приема лекарств любого типа - за исключением, конечно, случаев, когда это необходимо для спасения жизни или ликвидации серьезной угрозы для здоровья матери или плода
- Алкоголь. Употребление алкоголя приводит к разнообразным врожденным нарушениям, выраженность которых зависит от количества употребляемого алкоголя - особенно на ранних стадиях беременности. Фетальный (т.е. поражающий плод) алкогольный синдром - тяжелейшее врожденное заболевание, порой несовместимое с жизнью.
- Никотин. Курение сигарет во время беременности приводит к отставанию ребенка в физическом развитии
- Воздействие токсических химических веществ
Зачастую, однако, в развитии врожденных пороков играет роль такой фактор, как наследственная предрасположенность: известно, что если у родителей или ближайших родственников наблюдались врожденные пороки развития, то риск родить ребенка со сходными дефектами повышается, то есть речь идет "о семейном накоплении" аномалий развития. Так, у женщины с врожденным пороком сердца шансы родить ребенка с дефектом развития сердечно-сосудистой системы несколько выше, чем у всех остальных женщин. Поэтому принято говорить не столько о просто врожденных, сколько о врожденных мультифакториальных пороках развития. Тем не менее, на большом статистическом материале показано, что повторный риск рождения ребенка с врожденным пороком развития невелик - в среднем 2-4%.
Приведем несколько примеров совместимых с жизнью врожденных мультифакториальных пороков развития
Дефект развития
Выхождение внутренних органов или глубоких тканей из полостей, обычно занимаемых ими, под кожу или в межмышечную клетчатку без нарушения целости покровов
Массаж, в случае его неэффективности - хирургическое лечение
Врожденный вывих и врожденная дисплазия тазобедренного сустава
Врожденная дисплазия тазобедренного сустава - недоразвитие тканей тазобедренного сустава, отсутствие соответствия между суставными поверхностями - состояние, предшествующее вывиху тазобедренного сустава
При дисплазии - применение различных ортезов (приспособлений для отведения бедер) у детей до года. При вывихе - вправление, наложение специальных ортезов в первые месяцы жизни. При безрезультатности такого лечения - хирургическая операция.
Незаращение верхней губы (заячья губа)
Несращение боковых частей верхней губы с ее средней частью. Может быть односторонним и двусторонним. Затрудняет сосание
Хирургическая операция в первые месяцы жизни
Незаращение неба (волчья пасть)
Незаращение верхней челюсти и твердого неба, в результате чего получается расщелина, соединяющая полости рта и носа. Вызывает нарушение питания (попадание пищи в дыхательное горло, в полость носа), дыхания и речи. Часто сочетается с расщелиной в верхней губе
Хирургическая операция и протезирование; диспансерное наблюдение (смена лечебных аппаратов) до 16 лет
Полидактилия - многопалость, наличие лишних пальцев на кисти или стопе. Наиболее частый из врожденных пороков развития; чаще всего встречается в форме шестипалости, обычно на одной конечности.
Врожденный порок сердца
Неправильное внутриутробное формирование перегородки сердца (например, незаращение межпредсердной или межжелудочковой перегородки) либо сохранение после рождения особенностей внутриутробного кровообращения (например, открытый боталлов проток)
При незначительных дефектах межжелудочковой перегородки по мере роста сердца относительный размер отверстия уменьшается - вплоть до полного спонтанного закрытия. В других случаях - хирургическое лечение
Хотелось бы еще раз подчеркнуть, что когда речь идет о врожденных пороках развития, вопрос "Кто виноват?" не только непродуктивен, но и вреден, потому что отвлекает внимание от главного вопроса - "Что делать?". Поговорим на эту тему.
Что делать, если вы планируете беременность
- мужчины и женщины, в чьих семьях уже встречалось то или иное наследственное заболевание, - даже если сами они не больны
- семьи, где уже есть дети, страдающие врожденными пороками развития
- семьи, в которых предыдущие беременности заканчивались выкидышами или мертворождениями
- супруги, состоящие в родстве (например двоюродные и троюродные братья и сестры)
- женщины старше 35 и мужчины старше 50 лет
- мужчины и женщины, в связи со своим родом деятельности, состоянием здоровья или по каким-то иным причинам подвергающиеся воздействию перечисленных выше тератогенных факторов
Во всех этих случаях мы настоятельно рекомендуем партнерам, планирующим беременность, посетить медико-генетическую консультацию. Специалисты-генетики составят родословную, определят риск рождения ребенка с наследственным заболеванием. Нынешний уровень развития медицинских технологий позволяет сегодня в случае неблагоприятного прогноза прибегнуть к искусственному осеменению спермой донора или оплодотворению донорской яйцеклетки. Кроме того, следует по возможности исключить или свести к минимуму воздействие тератогенных факторов.
Что делать, если вы ждете ребенка?
Если вы ждете ребенка и входите в одну из перечисленных "групп риска". Первым шагом и в этом случае должен быть визит в медико-генетическую консультацию. Говорить об этом невесело, но бывают - хотя и очень редко - ситуации, когда на основании одной только родословной генетики приходят к заключению, что плод поражен заболеванием, несовместимым с жизнью. В таком случае, конечно, рекомендуется прерывание беременности. Однако, повторимся, случаи эти очень и очень редки. Как правило, специалисты медико-генетической консультации занимаются не диагностикой, а оценкой риска рождения ребенка с тяжелыми аномалиями и на основании этой оценки рекомендуют тот или иной метод пренатальной диагностики . Далее решение принимается в зависимости от результатов исследования. Насколько на самом деле высок риск родить ребенка с пороками развития, может решить лишь специалист. Не торопитесь делать аборт, если вы прочитали в аннотации, что лекарственный препарат, который вы принимали в самом начале беременности, не рекомендуется использовать в этот период; если принимали алкоголь, наркотики или перенесли острую респираторную вирусную инфекцию, сделали рентгеновский снимок на фоне беременности и т.п. Обязательно обратитесь в медико-генетическую консультацию, где сумеют правильно оценить реальный риск и порекомендуют необходимый комплекс исследований.
Что делать, если у вас родился ребенок с врожденным пороком развития
За окончательной медико-генетической консультацией по поводу прогноза на будущее лучше обратиться через 2-3 месяца, когда спадет психологическая напряженность и супруги смогут более объективно воспринимать такого рода информацию. Для большинства семей последующие беременности бывают успешными. Возможности пренатальной диагностики добавляют уверенности в благополучном исходе и врачам, и пациентам.
Имеются противопоказания. Ознакомьтесь с инструкцией или проконсультируйтесь у специалиста.
Революционные изменения в экспертной диагностике. Безупречное качество изображения, молниеносная скорость работы, новое поколение технологий визуализации и количественного анализа данных УЗ-сканирования.
Сцепленное с полом рецессивное наследование
Пороки развития, сцепленные с полом, в основном обусловлены рецессивными мутациями в женской половой хромосоме (этот тип наследования называют еще Х-хромосомным). Такой признак всегда передается через мать - носительницу рецессивного "больного" гена (т.е. сама женщина здорова). Практически все пораженные - мужчины (у пораженного гена Х-хромосомы в Y-хромосоме отсутствует "партнер", который мог бы доминировать над ним). Больной мужчина никогда не передает заболевания своим сыновьям (ведь они получают от него "здоровую" Y-, а не мутантную Х-хромосому), однако все его дочери будут носительницами "рокового" гена.
Мы схематически описали типы наследования, чтобы дать читателю общее представление о сути этих механизмов. На самом деле все гораздо сложнее - куда менее однозначно и определенно.
В приведенной ниже таблице перечислены в качестве примера лишь некоторые из совместимых с жизнью наследственных аномалий
Механизм наследования
Лечебно-реабилитационные меры
Аутосомно-рецессивное наследование - возможно рождение ребенка-альбиноса от здоровых родителей. Частота в популяции 1:20 000
Отсутствие нормальной пигментации кожи, волос, радужной оболочки глаза
Эта наследственная аномалия не считается заболеванием в полном смысле этого слова и лечению не подлежит
Сцепленное с полом рецессивное наследование. Болеют главным образом мужчины. Передается от матери сыновьям
Заболевание обусловлено дефицитом некоторых факторов свертывания крови. Проявляется кровоточивостью
Лечение при кровотечении - переливание крови, плазмы; кровоостанавливающие средства общего действия; антигемофильный глобулин; профилактика травм и кровотечений
Сцепленное с полом рецессивное наследование. Наблюдается преимущественно у мужчин. Передается от матери сыновьям
Частичная цветовая слепота. Распространяется чаще всего на красный и зеленый цвета
Расстройство цветового зрения выявляют при помощи специальных таблиц или спектральных приборов. Дальтонизм лечению не подлежит
Хромосомная аномалия: у матери при созревании яйцеклетки под влиянием пока не выясненных причин в 21-й паре хромосом образуется 3 хромосомы вместо 2-х. Частота в популяции - 1:700
Одна из форм врожденного слабоумия. Степень психического недоразвития значительно колеблется. Больные в основном ласковы, добродушны, приветливы
Лечебная педагогика, основанная на склонности больных к подражательности. Обучение во вспомогательных школах, трудотерапия
Аутосомно-доминантное наследование, передается детям от родителей с врожденной формой заболевания
Опущение верхнего века вследствие недоразвития мышцы, поднимающей его
Введение
Синдром ЕЕС (ectrodactyly-ectodermal dysplasia-clefting syndrome) - редкий генетический синдром, обычно проявляющийся триадой признаков: расщелиной лица, эктродактилией конечностей и признаками эктодермальной дисплазии [1]. Выделяют 2 типа синдрома: ЕЕС-1 и ЕЕС-3. Ранее был описан также и ЕЕС-2, однако на сегодняшний день локализация гена, кодирующего ЕЕС-2, считается ошибочной, следовательно, и формы ЕЕС-2 не существует. Первый тип характеризуется мутацией в 7-й хромосоме в области 7q11.2-q21.3, третий - имеет в большинстве случаев мутацию Тp63-гена и другие новые, спонтанные мутации на 3-й хромосоме 3q27 [2]. В OMIM (ежедневно обновляемом международном классификаторе Online Mendelian Inheritance in Man), где представлены все известные на сегодняшний день фенотипы и генотипы менделирующих (наследуемых) болезней человека, эти виды синдрома кодируются по-разному: ЕЕС-1 - 129900, а ЕЕС-3 - 604292 [3].
ЕЕС-синдром - наследственная патология с аутосомно-доминантным типом наследования, характеризующаяся различной пенетрантностью (проявляемостью) и экспрессивностью (степенью выраженности) [1].
Клиническое наблюдение 1
Пациентка Н. 24 лет. Беременность первая. Женщина и муж соматически здоровы, брак неродственный. Обратилась в медико-генетическое отделение МОНИИАГ в 21,4 недели беременности в связи с подозрением на порок развития конечностей у плода. Были выявлены: эктродактилия кистей (рис. 1) и стоп плода (рис. 2.). Из особенностей строения выявлены двусторонние пиелоэктазии (рис. 3). Проведено медико-генетическое консультирование. Семья приняла решение прервать данную беременность в связи с наличием инвалидизирующих пороков конечностей. При патологоанатомическом исследовании ВПР конечностей подтверждены, а также дополнительно выявлена расщелина мягкого неба и признаки эктодермальной дисплазии (гипертрофия десен, характерный лицевой фенотип). Окончательный диагноз: "синдром ЕЕС, аутосомно-доминантный тип наследования". Мутация de novo.
Экспертный класс по доступной цене. Монокристальные датчики, полноэкранный режим отображения, эластография, 3D/4D в корпусе ноутбука. Гибкая трансформация в стационарный сканер при наличии тележки.
Настоящая работа посвящена изучению плодового фенотипа при ультразвуковом исследовании в различные сроки беременности при помощи новейших технологий Samsung, оптимизирующих диагностику многих врожденных пороков развития, аномалий, нарушений строения, особенностей развития с оценкой значимости отдельных ультразвуковых патологических маркеров в качестве потенциальных признаков генетических синдромов и ассоциаций.
Одной из ведущих причин детской и младенческой смертности являются врожденные пороки развития (ВПР), среди которых в 20% случаев встречаются множественные ВПР (МВПР) с установленной частотой 1:250 новорожденных [1]. Особое значение в профилактике МВПР имеет пренатальная диагностика - современный и высокоэффективный метод диспансеризации плода, направленный на своевременное выявление патологии, впоследствии определяющий выбор адекватной акушерской тактики при данной беременности и специфических мер профилактики болезни в данной семье в дальнейшем [2]. Современные ее возможности позволяют из комплекса патологических нарушений у плода выделить достоверно значимые ультразвуковые маркеры для постановки диагноза генетических синдромов различной этиологии. Сегодня пренатальная синдромология активно развивается, как в плане расширения спектра возможных лабораторных исследований, так и описания значимых, ключевых, таргетных ультразвуковых признаков многих наследственных синдромов различного происхождения.
Группа МВПР включает сотни нозологических форм, различных по этиологии, фенотипическим проявлениям и прогнозу. В силу огромной гетерогенности МВПР представляют собой сложную проблему для постановки окончательного диагноза. Основными этиологическими факторами, приводящими к возникновению синдромов с множественными пороками развития, являются генные, хромосомные мутации и действие на плод неблагоприятных факторов внешней среды [3, 4].
Любые наследственные или врожденные генетические болезни являются результатом повреждения генетической информации на геномном, хромосомном или генном уровне. В 60% всех случаев МВПР встречаются хромосомные синдромы, достаточно широко изученные и хорошо диагностируемые. Для лабораторной диагностики этих повреждений разработаны разнообразные и хорошо известные методы: цитогенетический, молекулярно-цитогенетический и молекулярно-генетический. МВПР нехромосомного генеза встречаются в 40% случаев. Среди них выделяют синдромы, ассоциации и неклассифицированные комплексы. Применительно к нехромосомным синдромам цитогенетическое исследование позволяет лишь отвергнуть хромосомную природу данного комплекса пороков развития, но не установить диагноз.
Сегодня имеется определенное количество публикаций о выявлении некоторых синдромов и ассоциаций при пренатальной эхографии [6, 7]. Однако дородовая идентификация нозологических форм нехромосомных синдромов пока не вошла в рутинную практику врачей в области пренатальной диагностики и носит случайный характер. В литературе отсутствуют четкие рекомендации о системном подходе к диагностике наследственных синдромов различной этиологии, нет диагностических алгоритмов и определенных схем взаимодействия врачей ультразвуковой пренатальной диагностики, генетиков, акушеров-гинекологов.
Использование эхографии делает возможным пренатальное выявление не менее 80% врожденных дефектов во II, III триместрах беременности и свыше 50% значимых нарушений анатомии плода уже в I триместре начиная со сроков 11-12 нед. Весь комплекс диагностированных пороков развития и различных микроаномалий в совокупности составляет фенотипический спектр синдрома, который состоит из «ядерных» (главных, ключевых) признаков и дополнительных аномалий, наличие которых не является обязательным для диагностики клинической нозологии синдрома.
Революцией в пренатальной ультразвуковой диагностике явилось появление объемной эхографии, которая, обладая такими качествами, как неинвазивность, безопасность и возможность многократного применения у одной пациентки, имеет высокую информативность в исследовании анатомии плода и изучении его фенотипа. При применении различных режимов объемной эхографии абсолютно очевидно их преимущество по сравнению с обычным сканированием. Детально можно изучить лицо плода (рис. 1-4) в различные сроки беременности, начиная со сроков первого пренатального скрининга в 11-14 нед, конечности плода, причем не только их наличие и положение (рис. 5, 6), но и состояние и количество пальцев (рис. 7-9) как на руках, так и на ногах. Также можно изучить позвонки плода (рис. 10), состояние твердого нёба (рис. 11, 12), строение наружного уха (ушной раковины) (рис. 13), состояние основных швов черепа и родничков, исключая их преждевременное закрытие при кранисиностозах (рис. 14, 15).

Эталон новых стандартов! Беспрецедентная четкость, разрешение, сверхбыстрая обработка данных, а также исчерпывающий набор современных ультразвуковых технологий для решения самых сложных задач диагностики.
Заключение
Сложности ранней диагностики связаны с невыраженными УЗ-проявлениями достаточно тяжелой патологии. Четкое соблюдение алгоритма скрининговых УЗ-исследований анатомии плода в 11-14 и 20-22 нед беременности позволяет диагностировать экстрофию мочевого пузыря. При выявлении экстрофии мочевого пузыря у плода необходимо проведение медико-генетического консультирования с оформлением пренатального консилиума. Требуется тщательное разъяснение родителям сути выявленной патологии, а также информация о частых послеоперационных отдаленных последствиях и осложнениях как физического, так и психогенного генеза.
Введение
Экстрофия мочевого пузыря (ЭМП) - редкий тяжелый врожденный порок развития нижних мочевых путей, характеризующийся отсутствием передней брюшной стенки и передней стенки мочевого пузыря, а также расхождением лонного сочленения (диастаз). Через дефект передней брюшной стенки пролабирует слизистая оболочка. В нижней части обнаженной слизистой оболочки расположены устья мочеточников, из которых постоянно выделяется моча. Пуповина впадает низко в верхний край дефекта передней брюшной стенки.
Экстрофийные пороки нижних мочевых путей делят на классическую экстрофию мочевого пузыря и экстрофию клоаки. У мальчиков экстрофия мочевого пузыря сочетается с расщеплением мочеиспускательного канала (эписпадия). Экстрофия мочевого пузыря у девочек сопровождается расщеплением клитора, расщеплением или отсутствием мочеиспускательного канала, а также спайками больших и малых половых губ [1, 2].
Впервые экстрофию мочевого пузыря, как врожденный порок развития, описал Schenk von Grafenberg в 1597 г. В 1780 г. Chaussier впервые использовал термин "экстрофия". Упоминания об экстрофии мочевого пузыря обнаружены на ассирийских табличках, сделанных 2000 лет до н. э. [7]. Частота экстрофии мочевого пузыря составляет 0,25-0,5 на 10 тыс. новорожденных. Соотношение классической экстрофии мочевого пузыря у мальчиков и девочек 3:1 [1, 8]. При экстрофии мочевого пузыря встречаются сопутствующие врожденные аномалии - паховые грыжи (у 56 % мальчиков, у 15 % девочек), крипторхизм (20 %), колоректальные аномалии (1,8 %) и др. [1, 2]. Экстрофия мочевого пузыря может входить в состав комплекса OEIS (omphalocele, extrophy, imperforate anus, spinal defects), OMIM 258040.
Пренатальная диагностика экстрофии мочевого пузыря возможна уже при первом скрининговом обследовании в 11-14 нед беременности, когда в норме полость мочевого пузыря начинает наполняться мочой. Визуализация мочевого пузыря обязательно входит в стандартные протоколы исследования пренатального скрининга.
Аутосомно-рецессивный тип наследования
Носитель патологического признака - рецессивный ген, содержащийся в аутосоме. При аутосомно-рецессивном механизме наследования ситуация выглядит парадоксально - у здоровых родителей вдруг появляется на свет ребенок с дефектами развития, порой тяжелейшими и даже несовместимыми с жизнью. Причина - носительство обоими супругами в скрытом состоянии мутантных рецессивных генов. При этом рождение больного ребенка не обязательно означает, что все следующие дети будут страдать тем же заболеванием. Так же, как и в аутосомно-доминантном типе, мальчики и девочки в равной степени подвержены заболеванию.
Эмбриопатогенез
Если повреждение мембраны происходит до того, как спускается уроректальная перегородка, тогда это приводит к вывороту нижнего уретрального тракта и части пищеварительного тракта с формированием клоакальной экстрофии. Генетические факторы, связанные с экстрофией мочевого пузыря, на сегодняшний день не выявлены, однако существует гипотеза, что потеря экспрессии р63 из-за полиморфизмов очередности у промотера является фактором риска развития экстрофии мочевого пузыря [1, 9].
Ниже представлен случай диагностики экстрофии мочевого пузыря в III триместре беременности.
Аутосомно-доминантный тип наследования
Носитель патологического признака - доминантный ген, содержащийся в аутосоме (неполовой хромосоме). При этом типе наследования невозможно рождение больного ребенка у здоровых родителей - хотя бы один из родителей страдает от того же заболевания. При этом мальчики и девочки в равной степени подвержены заболеванию. Такие дефекты развития, как правило, бывают негрубыми и после успешной коррекции не препятствуют нормальной жизни.
Материал и методы
Пациентка С.,18 лет, настоящая беременность первая, от первого брака, супруги соматически здоровы, профессиональных вредностей не имеют, наследственность не отягощена. На учете в женской консультации пациентка состояла с 9 нед беременности. Беременность протекала без осложнений. При скрининговом УЗ-исследовании в I и II триместрах по месту жительства ВПР и УЗ-маркеров хромосомных перестроек у плода не обнаружено.
При УЗ-обследовании в 34 нед беременности в женской консультации по месту жительства было обнаружено образование средней эхогенности размером 28x23x25 мм в области нижних отделов передней брюшной стенки. Женщина была направлена в медикогенетическое отделение МОНИИАГ с диагнозом: "ВПР плода, экстрофия мочевого пузыря?".
УЗ-исследование проводилось на сканере WS80A (компании Samsung Medison) с использованием объемного датчика CV1-8A.
Читайте также:
- Цихлазома Сальвини или Манга-Цихлида: секреты содержания, ухода и разведения
- Болезни поросят и их симптомы, признаки, лечение, красные пятна на теле: что это
- Бенгальскому тигру изготовят протез для конечности
- Черный гриф попал на фотоловушку в приморье
- Миокардоз у животных: этиология, патогенез, клиническая картина, патологоанатомические изменения, диагноз, лечение, профилактика.